



The format for running STAMP is:
stamp -l <listfile> -s -o <output file> -P <parameter file>
-n <1 or 2 fits> -d <database file for scans>
-slide <slide value>
-pen1 <gap penatly 1> -pen2 <gap pentalty 2>
-prefix <output file prefix>
-V
-rough
-cut
-<parameter> <value>
If you have old STAMP parameter files, they can be read by using
the command stamp -P <parameter file>. This means that the old
file can be read in exactly the same way as for version 2.0.
In general, all commands can be specified by -<parameter> <value>.
For example, `-first_pairpen 0.5'. However, I have made some
abbreviations for frequently used commands, these are:
-l <list file> same as -listfile <list file> -o <output file> same as -logfile <output file> -n <1 or 2> same as -npass <1 or 2> -pen1 <gap penalty 1> same as -first_pairpen <gap penalty 1> -pen2 <gap penalty 2> same as -second_pairpen <gap pentalty 2> -prefix <output prefix> same as -transprefix <output prefix> -s same as -scan true -d <database file> same as -database <database file> -slide <slide parameter> same as -scanslide <slide parameter> -cut same as -co true -rough same as -roughfit true
Default parameters are always looked for in the file
STAMPDIR/stamp.defaults. You can personalise this is as you like, but I
would recommend using the defaults, unless you have a thorough
understnading of the method. The values described below were essentially
chosen to mimick the successful and well-tested parameters
[Russell & Barton, 1992].
I would recommend using the command line parameters. The commands, and
their arguments are given below. The command line parameters are case
insensitive. To use a parameter one need only type `-<parameter> <value>'
or use on of the short forms listed above.
STAMP can also be supplied with a parameter file. Parameters in
a parameter file can be supplied in the format:
<Parameter> <Value> <Optional Comments> [return]
eg.
PAIRWISE Yes Perform pairwise calculations E1 3.8 E2 3.8 CUTOFF 4.5
Since the program is written in C, the input is read in an open
format. Generally, data are expected to be separated by spaces or
return characters. The number and position of spaces, tabs and
returns generally should not matter with the exception of PDB
format, which is read as the fixed format described in the
brookhaven documentation.
The possible parameters are listed below. Strings, characters,
floats and integers are as expected (though strings may not contain
spaces). Boolean variables may be set by any of the following:
TRUE == TRUE, True, T, true, Yes, YES, yes, Y, 1 FALSE == FALSE, False, F, false, No, NO, n, 0
LOGFILE <string>
This is the file into which the log is to be written. If
`stdout' is supplied then the information is written to the
standard output.
Default LOGFILE = stdout
LISTFILE <string> (or `-l <string>' or `-f <string>')
This is the name of a file that contains the location and
description of the domains to be analysed and, if desired, an
initial transformation.
Default LISTFILE = domain.list
SEC <integer>
This must be set to 0 (no secondary structure assignment) or
to one of the following values:
SEC = 1 Kabsch and Sander's DSSP output. This program, which
calculates secondary structure based on hydrogen bonding criteria
[Kabsch & Sander, 1983] is available from the EMBL fileserver.
SEC = 2 Richards and Kundrot DEFINE output. This program, which
calculates secondary structure based on local distance criteria
[Richards & Kundrot, 1988], is available from Fred Richards. The required
format is from an adapted version of the program written by RBR. If you
are interested in making use of these definitions, you must acquire
the program, then refer to RBR for details on how to interpret the
output (ie. it is a bit finicky).
SEC = 3 Secondary structure summary format. A string of residue by
residue secondary structure assignments for each domain is to be
read in from SECFILE in the format specified in the previous chapter.
Note that it is not possible to mix assignments. This is probably
not a very realistic thing to do anyway, since assignments can
differ substantially. If you really want to do this, then the only
possible way is to set SEC = 3, and define each secondary structure
independently in SECFILE.
Default SEC = 1 (for DSSP).
SECFILE <string>
The file from which user specified secondary structure assignments
are to be read (ie. SEC = 3 only).
Default SECFILE = stamp.sec
PAIRWISE <boolean>
If TRUE, then pairwise comparisons are to be performed for each
possible pair of domains described in LISTFILE. A matrix of
pairwise (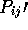 ) scores will be output (to MATFILE).
) scores will be output (to MATFILE).
Default PAIRWISE = TRUE
N.B. Many of the following parameters also apply to TREEWISE and
SCAN comparisons. For clarity they are discussed here in the
PAIRWISE comparison context.
NPASS <1 or 2> (or `-n <1 or 2>')
Whether one or two fits are to be performed. The idea is that the
initial fit can be used with a conformation biased set of
parameters to improve the initial fit prior to fitting using
distance and conformation parameters. The parameters described
below are called `first_' and `second_' accordingly. When NPASS =
1, then only the `second_' (or unprefixed) parameters are used.
Default NPASS = 1
SW <0 or 1>
If set to 0, then the entire M x N matrix will be calculated and
used during the Smith Waterman path finding routine. If set to 1,
then a corner cutting routine will be used (to save time). Note
that corner cutting will nullify many of the parameters specified
in [Russell & Barton, 1992], and is only recommended for SCAN mode.
Accordingly, corner cutting parameters are specified below (after
SCAN).
PAIRPEN <float> (or `-pen1 <float>'/ `-pen2 <float>')
(first_PAIRPEN)
(second_PAIRPEN)
Smith-Waterman gap penalty to be used during the fitting.
second_PAIRPEN and PAIRPEN are equivalent. (PAIRPEN is also
relevant to treewise fitting)
Defaults PAIRPEN = second_PAIRPEN = 0.0 first_PAIRPEN = 0.0
E1 <float>
E2 <float>
(first_E1,first_E2)
(second_E2,second_E2)
Rossmann and Argos parameters to be used during the fitting.
Rossmann and Argos suggested that E1 = E2 = 3.8 lead to good
superimpositions, and further suggested that E1 = 20.0 and E2 = 3.8
would relax the distance requirement, and allow poor initial
superimpositions to be improved. The defaults are defined
accordingly.
Defaults:
E1 = second_E1 = 3.8
E2 = second_E2 = 3.8
first_E1 = 20.0
first_E2 = 3.8
I would not recommend modifying these parameters, since I really
don't know what changing them will do. If it ain't broke, don't
fix it as my father would say.
NA <float>
NB <float>
NASD <float>
NBSD <float>
NSD <float>
NMEAN <float>
Parameters used to define 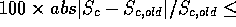 and
and 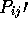 values. These are
defined in [Russell & Barton, 1992]. I wouldn't change these.
values. These are
defined in [Russell & Barton, 1992]. I wouldn't change these.
Defaults:
NA = -0.9497
NB = 0.6859
NASD = -0.4743
NBSD = 0.01522
NMEAN = 0.02
NSD = 0.1
CUTOFF <float>
(first_CUTOFF)
(second_CUTOFF)
This is the minimum 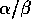 value allowed for atoms to be used
for a least squares fit. Equivalences above this value will be used to
determine a transformation and RMS deviation.
value allowed for atoms to be used
for a least squares fit. Equivalences above this value will be used to
determine a transformation and RMS deviation.
Defaults:
CUTOFF = second_CUTOFF = 4.5
first_CUTOFF = 1.0
PAIRALIGN <boolean>
If true, then each final pairwise alignment will be output to the
log file.
Default PAIRALIGN = FALSE
COLUMNS <integer>
Number of sequence positions to be displayed per line when either
PAIRALIGN, SCANALIN or TREEALIGN is set to TRUE.
Default COLUMNS = 80
SCORETOL <float>
This is the percent Sc difference that will result in convergence
being reached. In other words, if 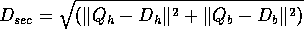 SCORETOL then
the fitting will be considered done.
SCORETOL then
the fitting will be considered done.
Default SCORETOL = 1.0
MAXPITER <integer>
The maximum number of iterations allowed during the pairwise
comparisons. This prevents a particular fit, which jumps between
two values rather than converging, from lasting indefinitely.
Default MAXPITER = 10
MATFILE <string>
This is the file which contains an upper diagonal matrix consisting
of the pairwise Scores (either 1/RMS, or Sc) for each comparison.
It may then be used to derive a tree, if desired, for treewise
analysis.
Default MATFILE = <stamp_prefix>.mat
ROUGHFIT <boolean> (or `-rough' to set to TRUE)
If set to TRUE, then an initial rough superimposition will be
performed by aligning the N-terminal ends of the sequences and
fitting on whatever atoms this process equivalences. Probably
this is too crude for structures that differ quite a bit, but if
they are very similar, one can use this to avoid having to
perform a multiple sequence alignment.
TREEWISE <boolean>
If TRUE, then a treewise comparison is performed by following a
derived hierarchy. Reads in the matrix file specified (either
created by PAIRWISE or some other method), derives a tree (dendrogram),
and does a tree-based alignment.
Default TREEWISE = TRUE
TREEPEN <float>
(first_TREEPEN)
(second_TREEPEN)
Value subtracted from the  matrix at positions where a
residue is to be aligned with a gap. For details see [Russell & Barton, 1992].
matrix at positions where a
residue is to be aligned with a gap. For details see [Russell & Barton, 1992].
Defaults TREEPEN = second_TREEPEN = 0.0 first_TREEPEN = 0.0
MAXTITER <int>
As for MAXPITER, but applied to the treewise case.
Default MAXPITER = 10
TREEALIGN <boolean>
As for PAIRALIGN, only for treewise comparisons.
Default TREEALIGN = TRUE
STAMPPREFIX <string> (or `-prefix <string>')
This is the name of the family of files that will be produced from
a multiple alignment. The files will be named STAMPPREFIX.<N>,
where N is the number of the cluster after which the alignment
has been derived. There are always one fewer clusters than their
are domains being compared.
Default STAMPPREFIX = `stamp_trans'
SCAN <boolean> (or simply `-s' to set true)
If TRUE, then SCAN mode is selected. TREEWISE and PAIRWISE are set
to FALSE. The first domain described in LISTFILE (the query) is
used to scan all the domains listed in DATABASE. The parameters
for scanning are described below. The output of a SCAN run appears
in the file called STAMPPREFIX.scan.
Default SCAN = FALSE
DATABASE <string> (or -d <string>)
The list of domains to be compared with the query during a scan.
Default DATABASE = domain.database
MAXSITER <int>
As for MAXPITER and MAXTITER, but for scanning. Equivalent
within the program to MAXPITER.
Default MAXSITER = 10
SCANALIGN <boolean>
As for PAIRALIGN and TREEALIGN, but for scanning. Equivalent
within the program to MAXPITER.
Default SCANALIGN = FALSE
SCANSCORE <integer>
Specifies how the Sc value is to be calculated. This depends on
the particular application. The values are described in the
first chapter.
As a general rule of thumb, use SCANSCORE=6 for large database
scans, when you are scanning with a small domain, and wishing to
find all examples of this domain -- even within large structures.
Use SCANSCORE=1 when you wish to obtain a set of
transformations for a set of domains which you know are similar
(and have defined fairly precisely as domains rather than the
larger structure that they may be a part of).
Default SCANSCORE = 6
SKIPAHEAD <boolean>
If set to TRUE, then the program will skip over all hits. In
other words, if a similarity is found with a particular starting
fit position, then the next fit position will be the last residue
of the similar region. This is not always desireable, since there can
be more than one hit within repetetive structures, such as  barrels.
barrels.
Default SKIPAHED = TRUE
SCANCUT <float>
If SCANMODE = 1, then Sc must be >= SCANCUT in order for a
transformation to be output.
Default SCANCUT = 2.0
SCANSLIDE <integer> (or `-slide <integer>')
This is the number of residues that a query sequence is `slid`
along a database sequence to derive each initial superimposition.
Initially, the N--terminus of the query is aligned to the 1st
residue of the databse, once this fit has been performed and
refined, and tested for good structural similarity, the N--terminus
is aligned with the 1+<SCANSLIDE>th position, and the process
repeated until the end of the database sequence has been reached.
Default SCANSLIDE = 5
SCANTRUNC <boolean>
If TRUE, then sequences from DATABASE that are more than
SCANTRUNCFACTOR x the length of the query sequence are truncated to
this size. This saves a lot of CPU time, as comparisons between
things that are vastly different in size are largely meaningless.
Moreover, since most scans will be done with discrete domains, then
this allows separate domains in large proteins to be compared
to the query separately.
Default SCANTRUNC = TRUE
SCANTRUNCFACTOR <float>
The largest size of sequence which may be compared to the query
sequence (expressed as SCANTRUNCFACTOR x query sequence length).
Structures in the DATABASE that are larger than this will be
truncated to this size if SCANTRUNC = TRUE.
Default SCANTRUNCFACTOR = 2.0
SLOWSCAN <boolean>
If set to TRUE, then the SLOW method of getting the initial fits
for scanning will be used (See chapter 1).
Default SLOWSCAN = FALSE
MIN_FRAC <float>
This is the minimum ratio of database length/query length to be
allowed. In other words, if a database structure is too small
(ie. if databaselength/query length < MIN_FRAC), then the
comparison will be skipped. Whether to use this or not depends on
whether or not one is interested in sub alignments where only a
part of the query structure is used. The default implies that all
comparisons will be performed.
Default MIN_FRAC = 0.001
SECSCREEN <boolean>
If TRUE, then an initial comparison between query and DATABASE
secondary structure assignments (if available) is performed. A
secondary structure distance is defined by:

where  and
and 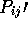 are the percent of Helix and Beta
structure in the query, and
are the percent of Helix and Beta
structure in the query, and 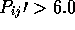 and
and 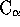 are the same for the
database sequence. If Dist is larger than a threshold
(SECSCREENMAX) then the comparison will be ignored.
are the same for the
database sequence. If Dist is larger than a threshold
(SECSCREENMAX) then the comparison will be ignored.
Default SECSCREEN = true
SECSCREENMAX <float>
This is the maximum value of Dist (above) tolerated. If Dist is
larger than SECSCREENMAX then the comparison is ignored. For
screening to be effective, it is important that secondary structure
assignments are accurate (preferably done using the same program).
Default SECSCREENMAX = 60.0 (this is very lenient; 40 is usually safe)
CCFACTOR <float>
Corner cutting factor. This is approximately the maximum number of
gaps to be tolerated in any pairwise comparison. Only used if SW = 1.
For a more detailed explanation, refer to [Sankoff & Kruskal, 1983] (pp
279 -- 281).
Default CCFACTOR = 30.0
CCADD <boolean>
If TRUE, then the difference between query and database sequence
lengths will be added to CCFACTOR. Probably this is only realistic
when SCANTRUNC is set TRUE.
Default CCADD = FALSE
PRECISION <integer>
Since STAMP works as much as possible with integers, this is what
all floating point values are multiplied by during conversion. A
value of 1000 has never presented us with any problems.
Default PRECISION = 1000
MAX_SEQ_LEN <integer>
The maximum length of alignment tolerated. The program ought to
inform you when this value is surpassed.
Default MAX_SEQ_LEN = 1500