



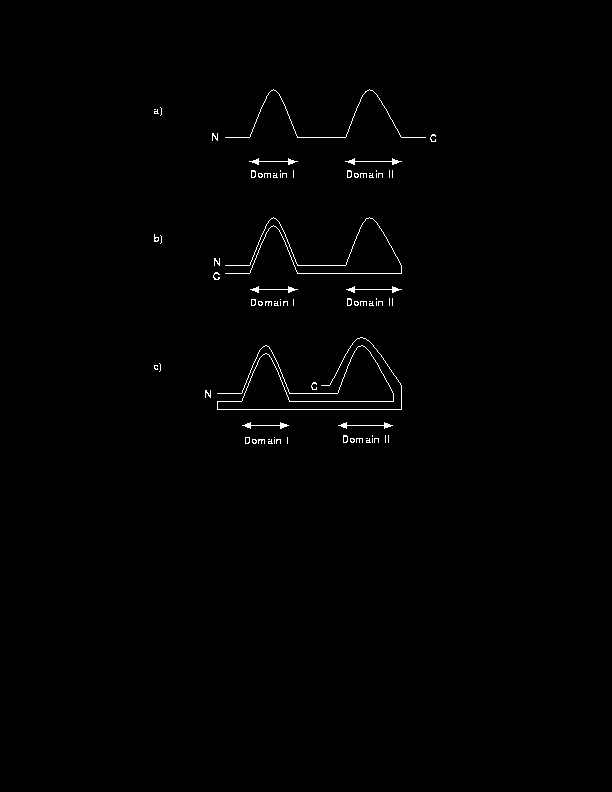
The concept of the domain has long been convenient to simplify and classify protein structure. Although there is no strict, universally accepted definition of a domain, domains are normally considered to be compact, local, semi-independent units [Richardson, 1981]. In a multi-domain protein, the domains may make up functionally and structurally distinct modules [Campbell & Baron, 1991,M Baron & Campbell, 1991]. Modules are usually formed from a single continuous segment of protein chain (Figure 1a) and it is conceptually easy to see how such domains with similar three dimensional structures may have arisen in different proteins by exon shuffling [Patthy, 1994]. However, examination of multi-domain proteins also reveals compact regions that are built of two or more non-sequential segments as illustrated in Figure 1b,c [Russell, 1994]. Although domains can be identified subjectively by eye, their importance to protein architecture and their possible role as independent nucleation sites in protein folding [Wetlaufer, 1973] prompted several groups during the late 1970s and early 1980s to investigate more systematic techniques for domain identification.
Rossmann and Liljas (1974) applied Phillips-Ooi - distance maps [Phillips, 1970,Nishikawa & Ooi, 1972,Nishikawa et al., 1972,Ooi & Nishikawa, 1973] to locate domains. They suggested that a domain has many short residue-residue distances within itself, but few short distances with the rest of the protein. Although a powerful abstraction, distance plots require human interpretation. In an attempt to automate the identification of domains, Crippen (1978) applied hierarchical cluster analysis to protein fragment/fragment contacts. This procedure generated a hierarchical tree of protein fragments from small, locally compact regions through to the complete protein. Rather than build up from fragments, Rose (1979) examined the complete protein to find the optimum point to cut the polypeptide chain based on the geometry of the protein. The procedure generated a hierarchy of fragments but was only able to deal with single segment (continuous) domains. Instead of considering cutting planes or simple distances, Wodak and Janin (1981) calculated the interface area between two segments of the protein. They chose the minimum in the interface area as the domain boundary. The approach was extended to deal with domains made of two segments, though this was computationally expensive and not fully automated. Rashin (1981) , Go (1983) and Zehfus and Rose (1986) applied globularity or compactness as domain definitions, but their methods could deal only with single segment domains. More recently, Zehfus (1994) used compactness as a measure of `domainness' and searched for compact units in the structure composed of two non-contiguous sections of the chain. The technique resulted in a series of overlapping domain units but did not provide a unique definition of the domains in the protein. Furthermore, the method could not be run in a reasonable time on proteins that contained more than 300 residues. Holm and Sander (1994) describe a method that searches for potential folding units using an eigenvalue analysis of contact maps. Although their elegant and fast method deals with multiple segment domains, many of their published domain definitions disagree with those found in the literature.
With the current rapid growth in the number of known protein three
dimensional structures, there is a pressing need to identify
systematically the domains. Knowledge of domain locations is
important in any reference database of protein structure, such
knowledge is also needed for construction of representative sets of
protein structures for derivation of parameters in prediction.
Prediction of protein structure by threading techniques
(Bowie & Eisenberg, 1993; D. T. Jones  1992; Bryant &
Lawrence, 1993; and for review Wodak & Rooman, 1993)
1992; Bryant &
Lawrence, 1993; and for review Wodak & Rooman, 1993)
is best approached at the domain level since this reduces the computational overhead. Furthermore, if effective methods are to be developed to identify domain boundaries in proteins of unknown three dimensional structure, then a reliable library of domains is required to derive the necessary parameters.
A problem faced by all methods of domain definition is how to assess the quality of the domains that are identified. The majority of the early techniques reviewed above apply a simple physical or geometric model to divide a protein into domains. Although domains defined in this way may provide new insights about the protein structure they do not always agree with the domain definitions in the literature. Accordingly, the approach adopted in this paper is to start from a subset of known protein structures for which the domain definitions have been well established, then derive a method that can reproduce the definitions automatically. The success of the method is evaluated by application to a larger test set of proteins. A domain reference set has been constructed from domain definitions described in the literature. Where definitions for a protein have not been described, assignments have been made by inspection. The new method starts from a simple geometric model similar to that used by Wodak and Janin (1981) (a domain has more residue-residue contacts within than without). However, alone this is insufficient to reproduce the normally accepted domain boundaries. The method has been refined to take into account secondary structure content and other factors in order to improve the agreement with the training set. Finally, three simple rules that are applied to any domain definition obtained by the method provide a ranking scheme to identify the definitions that are most likely to be correct.
The method explicitly allows for two segment domains and implicitly allows the formation of three or more segment domains. It runs in a reasonable time on proteins of any size and can optionally provide a hierarchical classification of compact regions within the protein.
A unique definition of the domains is presented for a set of 230 protein chains. Automatic screening of this set picked out 173 proteins of which 97% agreed with the reference definitions.
{kind=link}