



It is often desirable to compare a particular domain or protein
structure to a database of known 3D structures in order that structurally similar
proteins may be found. The PAIRWISE option within STAMP was a
logical starting point from which protein structure database scans
could be performed.
Given a single protein domain (a query) and a list of domains to which it is
to be compared (a database), STAMP can be used to perform all possible
comparisons of the query to the database structures. The initial
superimposition problem is solved by attempting more than one
initial fit with each database structure. This can be done in one of two ways,
which I will call FAST and SLOW, for the obvious reasons.
In FAST mode, fits are performed by lying
query sequence onto the database structure starting at every ith position,
where i is an adjustable parameter usually set to five (i.e. the sequence
is laid onto the 1st, 6th, 11th, etc. position). Diagramatically, this looks
like:
Q=query, D=database
Fit 1 Q -------
D -----------
Fit 2 Q -------
D -----------
Fit 3 Q -------
D -----------
<etc.>
This approach is fine if the query is a single domain, and there is a strong similarity in
the database structure. However, if similarity is weaker, or if the query is multi-domain (not
always a good idea, I would recommend splitting the structure into domains first, though this
may not always be possible), then SLOW mode will perform more fits (hence ``SLOW'') by sliding
query and database sequences along each other like:
Q=query, D=database
Fit 1 Q -------
D -----------
Fit 2 Q -------
D -----------
Fit 3 Q -------
D -----------
<etc.>
Fit N-2 Q -------
D -----------
Fit N-1 Q -------
D -----------
Fit N Q -------
D -----------
In this approach, initial superimpositions are calulated using many more fractions of query and
database structure, making detectiong of weak similarities more likely.
The residues that are
equivalenced by either FAST or SLOW procedures are used to perform an initial fit, which is
refined by the conformation-based and distance-based fit used during
PAIRWISE/TREEWISE comparison of distantly related structures. If a
high enough similarity score (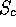 ) is found after these three
steps, then the transformation is saved for further analysis.
The output from the SCAN routine is directly readable by STAMP so
that once a list of domains similar to one's query is obtained,
multiple alignment (ie. PAIRWISE and TREEWISE) may be performed.
) is found after these three
steps, then the transformation is saved for further analysis.
The output from the SCAN routine is directly readable by STAMP so
that once a list of domains similar to one's query is obtained,
multiple alignment (ie. PAIRWISE and TREEWISE) may be performed.
The program PDBC can be used to generate a list of protein domains
given a set of PDB identifier codes, and the program SORTTRANS can
be used to sort the output from SCAN, and remove any redundancies.
The Sc values output during a SCAN differ slightly from those
output during a PAIRWISE comparison. The correction introduced
to correct the SW Score according to the length of the sequence
lengths is removed. During multiple alignment the start and end
points of the domains to be superimposed should be known, thus one
can penalise all for all positions which are not involved in the
alignment. During a scan, however, it is desireable to detect sub
alignments of the two structures being compared (eg., N-terminal helix
from query missing in database structure, a much longer database
structure, etc.). Thus, the Sc for scanning may be defined in one of
three ways (a=query, b=database, p=path, i=insertion, L=length):
Scheme 1
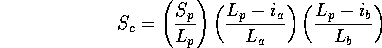
As for multiple structure alignment. As discussed, this is generally not the
best way to compare a
query to the database, since one would not usually wish to penalise insertions
or omitted missing segments within the database structure (due to truncation values,
etc.). However, this scheme may be useful if one is scanning a database of
structures known to exhibit a particular fold (i.e., if one is merely after
accurate superimpositions for a family of known structures; see Chapter 2).
Scheme 2
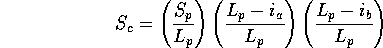
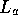 and
and 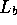 have been replaced by
have been replaced by 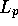 to removed any dependence
on query or database structure length. The second two terms
lower the score if gaps in the path are placed in query (a) or
database structure (b). This avoids a consideration of length, but will
allow short stretches structural equivalences to score highly.
to removed any dependence
on query or database structure length. The second two terms
lower the score if gaps in the path are placed in query (a) or
database structure (b). This avoids a consideration of length, but will
allow short stretches structural equivalences to score highly.
Scheme 3
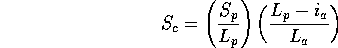
Only penalises insertions in the query sequence. If a small
fraction of the query sequence is in the actual path, then
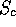 drops.
This scheme is most useful if one only wants similarities
to the entire protein under consideration, since it penalises
any omissions from the query structure.
drops.
This scheme is most useful if one only wants similarities
to the entire protein under consideration, since it penalises
any omissions from the query structure.
Scheme 4
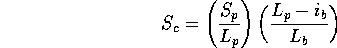
The opposite of 3. Only penalises insertions in the database sequence.
If a small fraction of the database sequence is in the actual path, then  drops. This scheme may be useful if one is scanning with a collection of
secondary structure elements, since gaps are to be expected within the
query (i.e., since the loops have been omitted).
drops. This scheme may be useful if one is scanning with a collection of
secondary structure elements, since gaps are to be expected within the
query (i.e., since the loops have been omitted).
Scheme 5

Raw score, no length requirement, will report even short alignments between
similar sub--structures. This scheme may be useful for the search for
short stretches of structural similarity, such as supersecondary structures.
Scheme 6
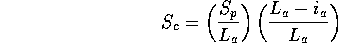
Vaguely similar to Scheme 3, but this only scores hits favourably
if the involve a significant fraction of the query structure
(i.e. similarities only containing part of the query will not
stand out). This is useful when one is comparing a particular
domain to a database and is not interested in local similarities.
This is the default for scanning.
For the most part, all of these scoring schemes will yield similar
numbers for very similar structures. However, when more distantly
related structures are compared, it becomes more useful to use a
scheme specific to the particular problem (i.e., whether one wishes
to scan with secondary structures only, when one is after only
very similar structures, etc.).
Schemes are specified by the parameter SCANSCORE (see below). If
you are confused, or haven't thought about this at all, just use the
defaults. I do.