



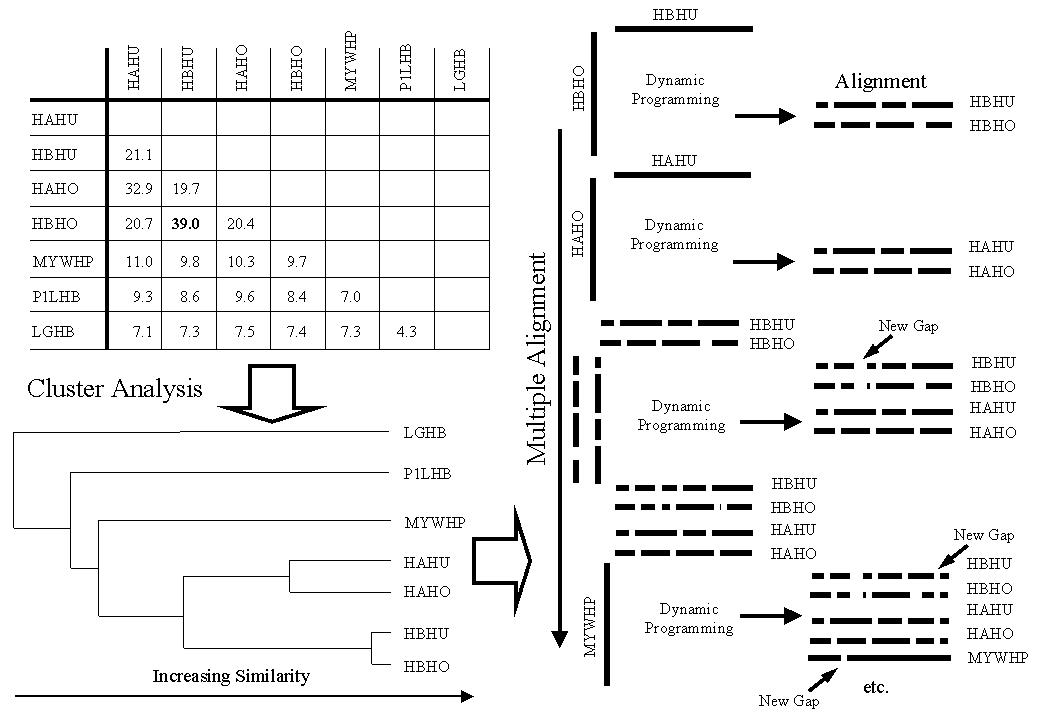
Hierarchical methods for multiple sequence alignment are by far the most commonly applied technique since they are fast and accurate. Hierarchical methods proceed in three steps. A schematic example of the stages in hierarchical multiple alignment is illustrated for 7 globin sequences in Figure 2.
For N sequences, all
![]() unique pair-wise comparisons
are made and the similarity scores for the comparisons recorded in
a table. The similarity scores, may be simple percentage identities,
or more sophisticated measures. For example, the SD score is
calculated by first aligning the pair of sequences by dynamic
programming and saving the raw score V for the alignment. The two
sequences are then shuffled and aligned, usually at least 100 times.
The score for each shuffled pair comparison is saved and the mean
unique pair-wise comparisons
are made and the similarity scores for the comparisons recorded in
a table. The similarity scores, may be simple percentage identities,
or more sophisticated measures. For example, the SD score is
calculated by first aligning the pair of sequences by dynamic
programming and saving the raw score V for the alignment. The two
sequences are then shuffled and aligned, usually at least 100 times.
The score for each shuffled pair comparison is saved and the mean
![]() and standard deviation
and standard deviation ![]() of the scores calculated.
The SD score is then given by:
of the scores calculated.
The SD score is then given by:
![]() .
SD scores
correct for the length and composition of the sequences, and so are
preferable to raw alignment scores, or percentage identity.
.
SD scores
correct for the length and composition of the sequences, and so are
preferable to raw alignment scores, or percentage identity.
In Figure 2, an example table of pair-wise SD scores is illustrated at the top left. Hierarchical cluster analysis is then performed on the table of pair-wise scores. This process generates a dendrogram or tree that groups the most similar pair, the next most similar pair and so on. The tree is illustrated at the bottom left of Figure 2. Finally, the multiple sequence alignment is generated by following the dendrogram from its leaves to the root. The detailed stages in this process are described in the legend to Figure 2.
One often stated drawback to hierarchical methods is that gaps once introduced are fixed. Thus, an error made at an early stage in the alignment process will propagate. Some methods allow a second pass through the alignment where each sequence is re-aligned to the complete alignment in order to correct the worst of these errors [Barton & Sternberg, 1987b,Gotoh, 1996].
The main differences between different hierarchical methods rest with the techniques used to score the initial pair-wise alignments, the hierarchical clustering method and the treatment of gaps in the multiple alignment. For example, pair-wise alignments may be scored by simple percentage identity, normalised alignment score (where the raw score for alignment of two sequences is divided by the length of the alignment) or a statistical SD score from Monte-Carlo shuffling of the sequences. Clustering may be a simple `add one sequence at a time' method [Barton & Sternberg, 1987b], or single linkage cluster analysis [Barton, 1990], or neighbour joining [Thompson et al., 1994], though any clustering method may be applied. Gaps in the multiple alignment process may be weighted simply, or position dependent [Thompson et al., 1994]. One of the most sophisticated hierarchical multiple alignment programs is CLUSTALW [Thompson et al., 1994]. CLUSTALW applies different pair-score matrices when aligning sequences of differing similarity. The program also modifies gap-penalties in a position specific fashion according to analyses of gap preferences in protein sequences [Pascarella & Argos, 1992].
{kind=link}