



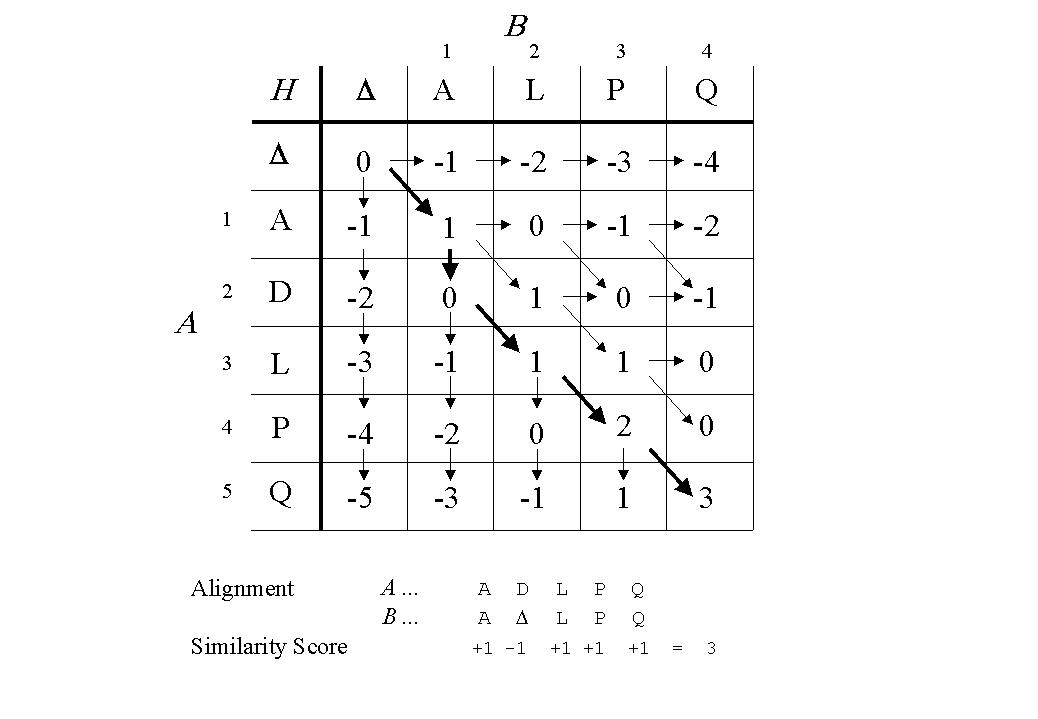
The optimal alignment of two protein sequences is the alignment that maximises the sum of pair-scores less any penalty for introduced gaps. The problem is to find an efficient way of locating an alignment that satisfies these conditions. If no gaps are allowed, then the task is simple, just slide one sequence over the other and for each position, sum the pair-scores from the chosen matrix (e.g. BLOSUM62). This simple alignment algorithm requires of the order of 200 alternative alignments to be evaluated for two sequences of length 100. In contrast, if gaps are allowed, then there are >1075 alternative alignments that must be considered! An elegant solution to this intractable search problem is given by a class of computer algorithm known as dynamic programming. Dynamic programming allows the optimal alignment of two sequences to be found in of the order of mnsteps, where m and n are the lengths of the sequences. Dynamic programming for sequence comparison was independetly invented in several fields, many of which are discussed in Sankoff and Kruskal's book [Sankoff & Kruskal, 1983]. An introduction to dynamic programming in the wider context of string comparison can be found in Gusfield (1997) . Needleman and Wunsch (1970) are often attributed as the first application of dynamic programming in molecular biology, while slightly different formulations of the same algorithm were described by Sellers [Sellers, 1974] and Waterman et al [Waterman et al., 1976]. Here, the basic Sellers [Sellers, 1974] dynamic programming algorithm for two sequences is described, with a length-dependent gap-penalty. A simple example of this algorithm is illustrated and explained in Figure 1.
At each aligned position between two sequences
![]() ,
and
,
and
![]() of length
m and n, there are three possible events:
of length
m and n, there are three possible events:
substitution of Ai by Bj (scores wAi, Bj),
deletion of Bj (scores
![]() ),
),
and deletion of Ai (scores
![]() ).
).
where ![]() is the symbol for a single residue gap. The
substitution score (
wAi, Bj) is taken from the amino-acid
pair-score matrix such as BLOSUM62 [Henikoff & Henikoff, 1992]. The two
deletion scores are the penalty for aligning a single residue
with a single gap.
is the symbol for a single residue gap. The
substitution score (
wAi, Bj) is taken from the amino-acid
pair-score matrix such as BLOSUM62 [Henikoff & Henikoff, 1992]. The two
deletion scores are the penalty for aligning a single residue
with a single gap.
The total score M for the alignment of A with B can be
represented as
![]() .
This is found by
working along each protein sequence from the N to C terminus,
successively finding the best score for aligning
.
This is found by
working along each protein sequence from the N to C terminus,
successively finding the best score for aligning
![]() with
with
![]() for all i, j where
for all i, j where
![]() and
and
![]() .
The values of
.
The values of
![]() are stored
in a matrix H where each element of H is calculated as follows:
are stored
in a matrix H where each element of H is calculated as follows:
Once the matrix is complete, the element Hm,n contains the total score for the alignment of the complete sequences.
The processing steps described above determine the best score possible for the alignment of the two sequences given the gap-penalty and pair-score matrix. However, they do not give the alignment. In order to generate an alignment of A and B that gives the best score, it is necessary to trace back through H. An efficient way of completing the trace-back is to save an array of pointers that records which of the three possibilities was the maximum at each Hi,j. It is possible for there to be ties for the maximum chosen at any Hi,j. Ties represent equally valid alternative alignments and the manner in which ties are resolved is dependent on the program implementation. For this reason, two computer programs that correctly claim to implement the same dynamic programming algorithm and give the same total score for the alignment of two sequences, may produce subtly different alignments.
Calculation of the best score is more complex if gap-penalties of the form ul + v are employed since it is necessary to determine for each cell whether a gap is starting at that cell, or continuing from an earlier cell. This algorithm typically runs a factor of 3 slower than the simple algorithm.
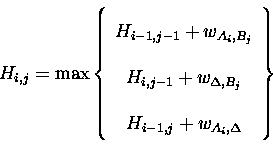
{kind=link}