



The ``All local alignment'' algorithm has been implemented in C. This language allows the C, M, S and E arrays to be grouped into a single data structure array of length m. The resulting code is faster than when C, M, S and E are coded separately, since sequentially accessed values are adjacent in memory. The results list is also of length m where each element points to a dynamically allocated ``ragged'' list of structures containing values for M, S and E.
As a check of the relative efficiency of the ``all local alignment''
algorithm, the protein sequence of human 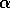 -- haemoglobin
(141-residues, PIR code HAHU) was compared to a small sequence
databank (PIR 14.0: 6,858 sequences, 2,080,148 amino acids) using the
Dayhoff MDM250 matrix [Dayhoff et al., 1978] and gap-penalty of 8
(
-- haemoglobin
(141-residues, PIR code HAHU) was compared to a small sequence
databank (PIR 14.0: 6,858 sequences, 2,080,148 amino acids) using the
Dayhoff MDM250 matrix [Dayhoff et al., 1978] and gap-penalty of 8
(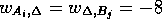 ). When run on a Sun
SPARCstation 2, the efficient ``best score only'' algorithm determined
one optimal local alignment for each databank sequence, and required
270 seconds to complete the scan. Another implementation of the
Smith-Waterman algorithm SSEARCH [] which also only
returns the top scoring alignment for each sequence pair, required
1,100 seconds for the same scan. In contrast, the ``all local
alignment'' algorithm described here with
). When run on a Sun
SPARCstation 2, the efficient ``best score only'' algorithm determined
one optimal local alignment for each databank sequence, and required
270 seconds to complete the scan. Another implementation of the
Smith-Waterman algorithm SSEARCH [] which also only
returns the top scoring alignment for each sequence pair, required
1,100 seconds for the same scan. In contrast, the ``all local
alignment'' algorithm described here with 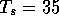 examined 30,690
local alignments in only 1,000 seconds.
examined 30,690
local alignments in only 1,000 seconds.
The value of 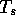 has little effect on the execution time, except
when set very small such that large numbers of local alignments must
be stored and sorted for each sequence comparison. For example,
comparison of HAHU (141 residues) to one of the longest protein
sequences known, Twitchin from Caenorhabditis elegans (PIR code
S07571 - 6048 residues), finds 32,299 alternative local alignments
when
has little effect on the execution time, except
when set very small such that large numbers of local alignments must
be stored and sorted for each sequence comparison. For example,
comparison of HAHU (141 residues) to one of the longest protein
sequences known, Twitchin from Caenorhabditis elegans (PIR code
S07571 - 6048 residues), finds 32,299 alternative local alignments
when 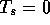 and requires 16 sec CPU time. Setting
and requires 16 sec CPU time. Setting 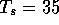 reduces the number of alignments to 58 and takes only 2 sec. The
algorithms described here have also been implemented to work with
pscan [Barton, 1991] to allow distributed processing on a network of
workstations. pscan permits an approximately linear decrease in
elapsed scan time with increasing numbers of processors.
reduces the number of alignments to 58 and takes only 2 sec. The
algorithms described here have also been implemented to work with
pscan [Barton, 1991] to allow distributed processing on a network of
workstations. pscan permits an approximately linear decrease in
elapsed scan time with increasing numbers of processors.