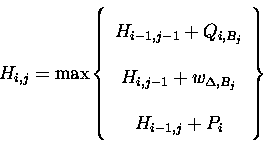In the examples in the previous sections, residue substitution weights have all come from a single pair-score matrix such as BLOSUM62. However, the substition weights ( wAi,Bj) can be made position-specific in the same way as gap-penalties. If the weights are position-specific relative to A it means that the weight for matching a residue from A of a particular type to any other residue will depend upon the location of the residue in A as well as the type of the residue in B. For example, a Gly at A32 aligning with a Gly in B may have a weight of +7, while a Gly at A76aligned with a Gly in B has a weight of -5. Position-specific weights are useful since they allow the importance of specific substitutions to be emphasised at particular positions along the sequence. This might mean increasing the weight for aligning a known active site residue in A with a residue of the same type, or more general properties such as increasing the weight for known buried hydrophobic residues to align with similar residues in B.
In order to calculate an alignment score by dynamic programming that
includes position-specific weights, a position-specific weight matrix
or profile Qm,20 for sequence A must be defined. Qcontains m rows where each row has 20 weights for substitions with
each amino acid type at that position. Thus, Q3,S is the
substitution weight for a Serine in B with position 3 of A. The
equivalent of having no position-specific weights is to populate each
![]() with the appropriate row from the BLOSUM62 or other
pair-score matrix. However, if the power of position-specific weights
is to be exploited, additional information about A must be included
in the derivation of Q. This might be knowledge of the three
dimensional structure of the protein with sequence A, where
position-specific weights reflect the local environment of the amino
acids (e.g Overington et al, 1992). Or
more commonly, the frequencies of observed amino acids at each
position in a multiple sequence alignment of sequences similar to A[Gribskov et al., 1987,Barton & Sternberg, 1987b,Barton & Sternberg, 1990].
with the appropriate row from the BLOSUM62 or other
pair-score matrix. However, if the power of position-specific weights
is to be exploited, additional information about A must be included
in the derivation of Q. This might be knowledge of the three
dimensional structure of the protein with sequence A, where
position-specific weights reflect the local environment of the amino
acids (e.g Overington et al, 1992). Or
more commonly, the frequencies of observed amino acids at each
position in a multiple sequence alignment of sequences similar to A[Gribskov et al., 1987,Barton & Sternberg, 1987b,Barton & Sternberg, 1990].
Hi,j when calculated with position-specific weights and gaps for A is modified to:
Since their parallel development by many authors in the mid 1980's [Gribskov et al., 1987,Barton & Sternberg, 1987b,Taylor, 1986b], position-specific weighting schemes, or profiles have formed the basis of many methods for sensitive sequence comparison. In addition, the principle of position-specific weights is at the heart of currently popular techniques such as Hidden Markov Models (HMM's) [Krogh et al., 1994] and generalised profiles [Bucher et al., 1996].
A generalisation of position-specific weight matrices is to create two Qmatrices, one for each sequence. When calculating each element of H, the substitution weights may be combined by averaging. Comparison of two profiles is fundamental to hierarchical multiple alignment algorithms discussed in Section 5.2.